

一、全国医疗器械不良事件报告总体情况
2013年,全国医疗器械不良事件报告工作呈现良好的发展态势。报告数量持续增长,已突破23万份,平均百万人口报告数已达179份;不良事件报告的整体质量有所提升;各地对不良事件报告的重视程度明显提高。
(一)2013年可疑不良事件报告数量
2013年,国家药品不良反应监测中心共收到《可疑医疗器械不良事件报告表》238,650份,与2012年相比增长31.7%。企业后续提交《医疗器械不良事件补充报告表》705份;《医疗器械不良事件年度汇总报告表》1,805份(表1)。
表1 2013年可疑不良事件报告数量与2012年比较情况
| 报告类型 |
报告数量 |
年度增长率(%) |
|
| 2012年 |
2013年 |
||
| 《可疑医疗器械不良事件报告表》 (《医疗器械不良事件补充报告表》) |
181255 (634) |
238650 (705) |
31.7 (11.2) |
| 《医疗器械不良事件年度汇总报告表》 |
1381 |
1805 |
30.7 |
1.死亡及严重伤害事件报告数量
2013年,国家药品不良反应监测中心共收到死亡不良事件报告75份,严重伤害事件报告34,524份,共计34,599份,占可疑不良事件报告总数的百分比为14.5%,比2012年的23,548份增长了46.9%。(图1)。
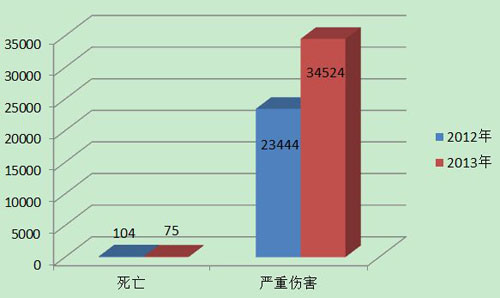
图1 2012-2013年全国死亡及严重伤害事件报告数比较
2.补充报告数量
2013年,国家药品不良反应监测中心共收到医疗器械不良事件补充报告705份,年度增长率11.2%,相对于死亡及严重伤害事件报告的快速增长,承载更多信息的补充报告数量增长缓慢,说明生产企业作为报告主体的主动性不够。各级监测机构对生产企业的督促还需加强,在监管同时加大技术指导力度,帮助企业做好监测和报告工作。(图2)。
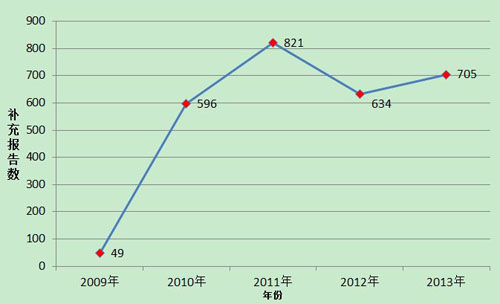
图2 2009-2013年全国医疗器械不良事件补充报告数量
3.年度汇总报告数量
按照《医疗器械不良事件监测和再评价管理办法(试行)》的要求,第二、三类医疗器械生产企业应当在每年1月底前对上一年度医疗器械不良事件监测情况进行汇总分析,并填写《医疗器械不良事件年度汇总报告表》,报所在地省、自治区、直辖市医疗器械不良事件监测技术机构。
2013年,国家药品不良反应监测中心共收到医疗器械不良事件年度汇总报告1,805份,仅占全国现有的二、三类医疗器械生产企业数量(二类8,255家,三类2,513家)的16.8%。
4.每百万人口平均报告数量
2013年,我国每百万人口平均可疑医疗器械不良事件报告数为179份,已经达到并超过《国家药品安全“十二五”规划》中100份/百万人的要求,但各省间发展不平衡,还有1/3的省份百万人口报告数偏低。2013年百万人口平均报告数与2012年相比增长了30.7%,与2011年相比增长了94.6%。较之报告总数的增长,百万人口报告数可更好地反映报告数量的增长情况(图3)。
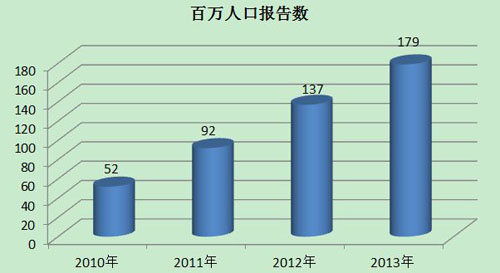
图3 2010-2013年全国百万人口报告数量比较
5.注册基层用户数量
截止到2013年12月31日,在全国医疗器械不良事件监测系统中,注册基层用户(包括生产企业、经营企业和使用单位)共148,585家。其中,医疗器械生产企业8,495家,约占注册基层用户总数的5.7%;经营企业61,498家,约占注册基层用户的41.4%;使用单位78,592家,约占注册基层用户的52.9%(图4)。
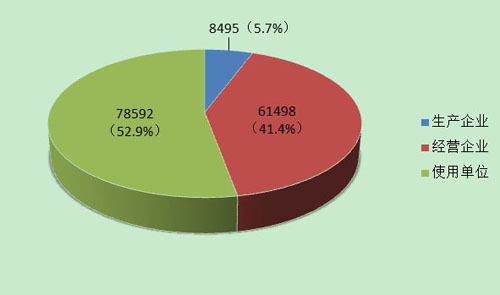
图4 2013年全国医疗器械不良事件监测系统基层用户注册情况
2013年,在148,585家注册基层用户中,提交过报告的单位数量为31,505家,占21.2%,基层用户上报情况详见表2。
表2 基层用户上报报告情况
| 基层用户 |
注册数 |
上报报告的单位数量 |
百分比(%) |
|||
| 总数 |
可疑医疗器械 不良事件报告 |
医疗器械不良 事件补充报告 |
医疗器械不良 事件年度汇总报告 |
|||
| 经营企业 |
61498 |
9576 |
9576 |
0 |
0 |
15.6 |
| 使用单位 |
78592 |
19335 |
19335 |
0 |
0 |
24.6 |
| 生产企业 |
8495 |
2594 |
975 |
77 |
1805 |
30.5 |
| 合计 |
148585 |
31505 |
29886 |
77 |
1805 |
21.2 |
(二)2002年至2013年全国可疑不良事件报告总数量
2002年1月1日至2013年12月31日,国家药品不良反应监测中心累计收到《可疑医疗器械不良事件报告表》735,559份(图5)。
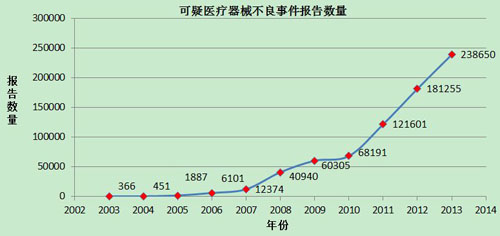
图5 2002-2013年全国可疑医疗器械不良事件报告数量
二、医疗器械不良事件报告统计分析
(一)按报告来源统计分析
2013年医疗器械不良事件报告中,使用单位上报175,317份,占报告总数的73.5%;生产企业上报7,274份,占报告总数的3.1%;经营企业上报52,736份,占报告总数的22.1%;还有3,323份报告来自于个人,占报告总数的1.4%(图6)。
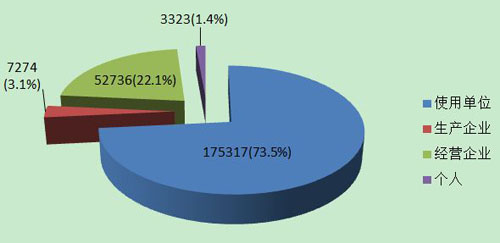
图6 2013年医疗器械不良事件报告来源
总体来看,来自于使用单位的报告较多,来自于生产企业的报告较少。相比2012年,生产企业的报告数绝对值及所占的百分比都有所上升,但仍与其第一责任人的地位不符。提示生产企业履行职责的自觉性还有待进一步提高。经营企业报告所占的百分比由2012年的21.58%上升至22.1%。
2013年,注册基层用户数量达到148,585,比2012年增加71.4%。但上报不良事件报告的单位所占比例与去年基本持平。在所有注册的基层用户中,仅有21.2%的用户提交过不良事件报告。其中,经营企业的上报比例最低,注册的61,498家用户中有9,576家提交了报告,仅占15.6%;生产企业注册的8,495家用户中有2,594家提交了报告,占30.5%;使用单位注册的78,592家用户中有19,335家提交了报告,占24.6%。尽管注册基层用户的数量大幅增加,但用户提交报告比例偏低,提示对基层用户的宣传培训、督促检查工作还需要进一步加强(图7)。
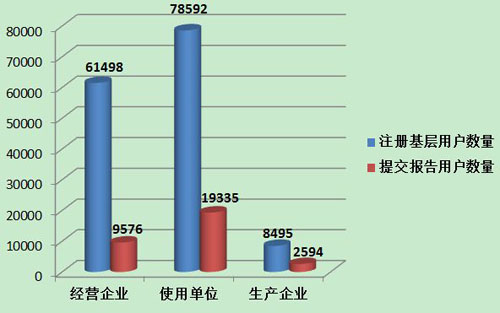
图7 2013年注册基层用户提交报告情况
(二)按事件伤害程度统计分析
2013年可疑医疗器械不良事件报告中,事件伤害为死亡的报告共75份,占总报告数的0.0003%;事件伤害为严重伤害的报告34,524份,占总报告数的14.5%;事件伤害为其他的报告共204,051份,占总报告数的85.5%(图8)。
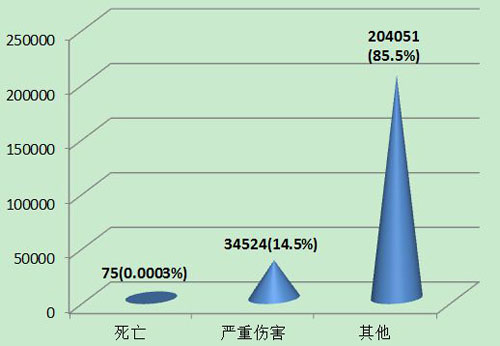
图8 2013年医疗器械不良事件报告事件伤害程度情况
2013年,严重伤害事件报告数量及所占比例均比2012年有所增加。总体来看,可疑医疗器械不良事件的报告数量延续了2009年以来的增长趋势,报告的质量及信息的可利用程度等有了较大提升(图9)。
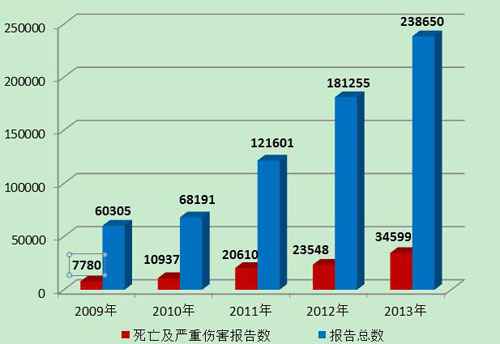
图9 2009-2013年全国报告数量与死亡及严重伤害报告数量比较
(三)按医疗器械管理类别统计分析
2013年医疗器械不良事件报告中,涉及Ⅲ类医疗器械的报告最多,共105,032份(其中死亡及严重伤害事件报告22,416份),占总报告数的44.0%;涉及Ⅱ类医疗器械的报告次之,共80,394份(其中死亡及严重伤害事件报告9,157份),占总报告数的33.7%;涉及Ⅰ类器械的报告最少,共48,254份(其中死亡及严重伤害事件报告2,597份),占总报告数的20.2%;部分报告涉及的器械管理类别不详,共4,970份报告(其中死亡及严重伤害事件报告429份),占报告总数的2.1%。Ⅱ类、Ⅲ类医疗器械的报告是构成主体,与医疗器械风险程度高低相吻合(图10,图11)。
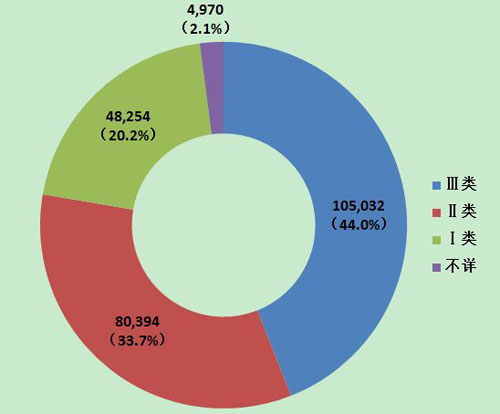
图10 2013年医疗器械不良事件报告涉及产品管理类别情况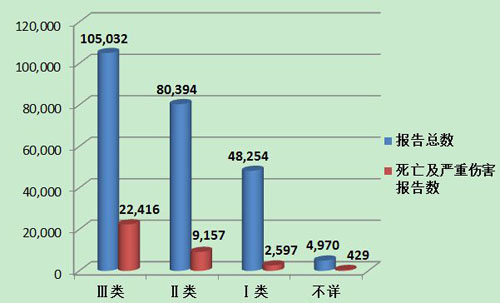
图11 2013年死亡及严重伤害医疗器械不良事件报告涉及产品管理类别情况
(四)按医疗器械分类产品目录统计分析
按照现行的《医疗器械分类目录》,2013年的可疑医疗器械不良事件报告共涉及43类产品(除产品分类不详或未填写等其他外),涵盖了《医疗器械分类目录》中的所有医疗器械类别。其中,报告数量位列前十位的产品类别依次为医用高分子材料及制品,注射穿刺器械,医用卫生材料及敷料,植入材料和人工器官,物理治疗设备,医用光学器具、仪器及内窥镜设备,普通诊察器械,医用电子仪器设备,手术室、急救室、诊疗室设备及器具和医用缝合材料及粘合剂。与2012年相比,报告数量位列前十位的产品类别完全相同,仅排名略有变化(表2)。
表2 2013年医疗器械不良事件报告涉及产品分类目录情况
| 序号 |
分类 |
报告数 |
构成比(%) |
| 1 |
6866医用高分子材料及制品 |
38413 |
16.1 |
| 2 |
6815注射穿刺器械 |
31589 |
13.2 |
| 3 |
6864医用卫生材料及敷料 |
30980 |
13.0 |
| 4 |
6846植入材料和人工器官 |
24912 |
10.4 |
| 5 |
6826物理治疗设备 |
24639 |
10.3 |
| 6 |
6822医用光学器具、仪器及内窥镜设备 |
13736 |
5.8 |
| 7 |
6820普通诊察器械 |
9695 |
4.1 |
| 8 |
6821医用电子仪器设备 |
7100 |
3.0 |
| 9 |
6854手术室、急救室、诊疗室设备及器具 |
6459 |
2.7 |
| 10 |
6865医用缝合材料及粘合剂 |
4830 |
2.0 |
(五)按可疑医疗器械不良事件报告数量排名前五位产品统计分析
2013年可疑医疗器械不良事件报告中,报告数量排名前五位的无源医疗器械分别为一次性使用输液器、宫内节育器、一次性使用无菌注射器、角膜接触镜和静脉留置针,占总报告数的34.0%,详见表3。报告数量排名前五位的有源医疗器械分别为病人监护仪、输液泵和注射泵、电子血压计、心电图机和呼吸机,占报告总数的4.3%,详见表4。
表3 报告数量排名前五位的无源医疗器械
| 序号 |
产品名称 |
报告数(n) |
占报告总数的百分比(%) |
严重伤害报告数 |
占本类产品报告数的百分比(%) |
| 1 |
一次性使用 输液器 |
28824 |
12.1 |
2148 |
7.5 |
| 2 |
宫内节育器 |
19822 |
8.3 |
9811 |
49.5 |
| 3 |
一次性使用无菌注射器 |
16517 |
6.9 |
823 |
5.0 |
| 4 |
角膜接触镜 |
8174 |
3.4 |
2201 |
26.9 |
| 5 |
静脉留置针 |
7857 |
3.3 |
1198 |
15.2 |
| 合计 |
81194 |
34.0 |
16181 |
- |
表4 报告数量排名前五位的有源医疗器械
| 序号 |
产品名称 |
报告数(n) |
占报告总数的百分比(%) |
严重伤害报告数 |
占本类产品报告数的百分比(%) |
| 1 |
病人监护仪 |
3761 |
1.6 |
742 |
19.7 |
| 2 |
输液泵和注射泵 |
2708 |
1.1 |
539 |
19.9 |
| 3 |
电子血压计 |
2033 |
0.9 |
60 |
3.0 |
| 4 |
心电图机 |
942 |
0.4 |
120 |
12.7 |
| 5 |
呼吸机 |
747 |
0.3 |
240 |
32.1 |
| 合计 |
|
10191 |
4.3 |
1701 |
- |
(六)按涉及使用人员统计分析
2013年可疑医疗器械不良事件报告中,70.1%的报告所涉及的医疗器械是由专业人员操作的;2.2%的报告所涉及的医疗器械是由非本人的非专业人员操作的;11.2%的报告所涉及的医疗器械是由患者自己操作的;16.5%的报告操作人不详。其中,由专业人员操作的报告所占比例与2012年相比基本持平(2012年,由专业人员操作的报告占报告总数的72.1%)。现有信息提示,操作人员是分析事件发生原因时要考量的重要因素之一,而报告操作人不详所占比例偏大,提示报告质量仍有待提高(图12)。
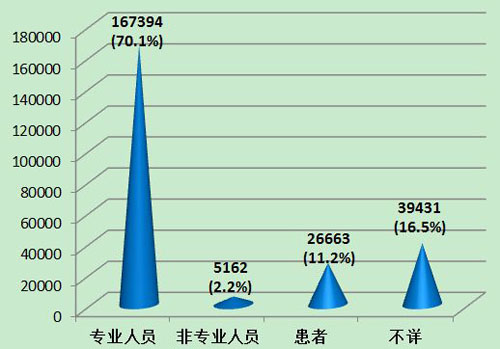
图12 2013年医疗器械不良事件报告涉及使用人员情况
(七)按涉及实际使用场所统计分析
2013年可疑医疗器械不良事件报告中,使用场所为“医疗机构”的报告161,871份,占67.8%;使用场所为“家庭”的报告共37,855份,占15.9%;使用场所为“其他”(如体验中心等)的报告共9,625份,占4.0%;使用场所为“不详”的报告共29,299份,占12.3%。医疗器械的使用场所呈现多元化的趋势,使用场所的复杂性也是分析不良事件发生原因时需要考量的因素之一(图13)。
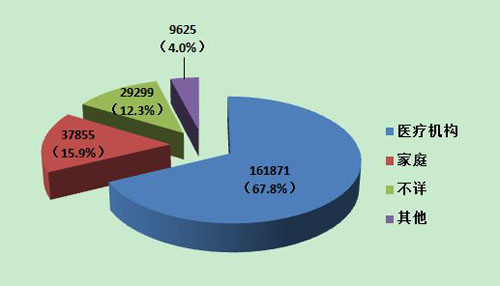
图13 2013年医疗器械不良事件报告涉及实际使用场所情况
三、对医疗器械安全性问题采取的主要措施情况
(一)发布《医疗器械不良事件信息通报》
2013年,共发布《医疗器械不良事件信息通报》3期,涉及接骨板、人工髋关节、医用电子直线加速器三个产品(表4-1)。
接骨板的临床使用风险主要表现为:钢板断裂、弯曲、松动,排异反应、异常疼痛、骨折不愈合、感染等。风险控制建议:生产企业应重视产品的不良事件监测,开展分析和再评价,进一步提高产品性能,加强质量控制,并对临床医务人员提供必要的技术支持和培训。临床医务人员应严格掌握手术适应症及禁忌症,选择适当规格型号的产品;严格按照说明书和手术规范的要求进行操作;③规范术前术后对病人的管理。
人工髋关节的临床使用风险主要表现为:假体松动、断裂、周围感染,关节脱位,术后疼痛,假体骨水泥植入综合征等。风险控制建议:生产企业应改进产品设计,进一步提高产品性能,提高生产工艺,选择生物相容性好、耐磨损、刚度适宜的材料,加强质量控制;提供详尽的标签及使用说明书,并对临床医务人员提供必要的技术支持和培训。临床医务人员应严格掌握关节置换的适应症及禁忌症;加强业务培训,严格按照说明书和手术规范的要求进行操作;规范术后护理;加强人工关节置换准入制度的管理,加强验收、储存、使用控制,建立详细的使用记录。
医用电子直线加速器的临床使用风险主要表现为:放射治疗引发的并发症(白细胞、血小板减少、脱发、放射性皮炎、非照射部位炎症、恶心、呕吐、腹胀、腹泻等。以及机械故障、电气故障、和软件故障等。风险控制建议:生产企业应完善售后产品技术支持,提高产品可靠性;加强对临床医务人员的培训;对常见设备故障,应与临床共同探讨,提出改进措施。医务人员应严格掌握放射治疗的适应征,按照相关规章制度及说明书要求维护、操作、使用电子直线加速器,制定科学合理治疗方案。
原文链接:
http://www.cdr.gov.cn/xxtb_255/ylqxblsjxxtb/
(二)发布《医疗器械警戒快讯》
2013年,共发布《医疗器械警戒快讯》10期,共71条安全性信息,涉及体外除颤仪、呼吸机、麻醉机、人工关节、血糖监测仪、腹膜透析管、电动轮椅、计算机断层扫描图像处理工作站等产品。
(三)召开产品专家分析会及产品企业沟通会
2013年,总局、国家中心共召开了4次医疗器械产品专家分析会、1次产品生产经营企业沟通会。
专家分析会涉及的产品为接骨板、人工髋关节、医用电子直线加速器和制氧机。会议对产品相关的安全性事件及可疑不良事件报告进行了讨论分析,并针对产品常见风险点提出了风险控制意见,采取了风险控制措施。
企业沟通会涉及的产品为壳聚糖。国家中心针对壳聚糖相关产品的不良事件监测情况及使用中存在的安全性问题组织召开了企业沟通会。生产企业代表介绍了其产品的概况及不良事件收集与处置情况。会议对壳聚糖类产品的风险及可能原因进行了分析,并提出了风险控制建议。
医疗器械不良事件监测小贴士
1.医疗器械:是指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,包括所需要的计算机软件;其效用主要通过物理等方式获得,不是通过药理学、免疫学或者代谢的方式获得,或者虽然有这些方式参与但是只起辅助作用;其目的是:
(1)疾病的诊断、预防、监护、治疗或者缓解;
(2)损伤的诊断、监护、治疗、缓解或者功能补偿;
(3)生理结构或者生理过程的检验、替代、调节或者支持;
(4)生命的支持或者维持;
(5)妊娠控制;
(6)通过对来自人体的样本进行检查,为医疗或者诊断目的提供信息。
2.医疗器械不良事件:指获准注册或已备案、质量合格的医疗器械,在正常使用情况下发生的,导致或可能导致人体伤害的各种有害事件。
根据医疗器械不良事件的危害程度和发生的原因,医疗器械生产企业必要时应当采取警示、检查、修理、重新标签、修改说明书、软件升级、替换、收回、销毁等控制措施。
目前,我国医疗器械不良事件监测按照“可疑即报”原则收集报告,即为可疑医疗器械不良事件报告。
3.医疗器械不良事件监测:是指对医疗器械不良事件的发现、报告、调查、评价和控制的过程。
4.严重医疗器械不良事件:指有下列情况之一者:
(1)导致死亡;
(2)危及生命;
(3)导致机体功能的永久性伤害或者机体结构的永久性损伤;
(4)必须采取医疗措施才能避免上述永久性伤害或者损伤;
(5)由于医疗器械故障、可用性等问题可能导致上述所列情况的。
5.医疗器械不良事件与质量事故、医疗事故的区别
(1)医疗器械不良事件主要是由于产品的设计缺陷、已经注册审核的使用说明书不准确或不充分等原因造成的,但其产品的质量是合格的。
(2)医疗器械质量事故主要是指其质量不符合注册产品标准等规定造成的事故。
(3)医疗事故是指医疗机构及其医务人员在医疗活动中,违反医疗卫生管理法律、行政法规、部门规章和诊疗护理规范、常规,过失造成患者人身损害的事故。(摘自卫生部《医疗事故处理条例》)





